Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming
- Martin Picarda,
- Jiangwen Zhangb,
- Saege Hancockc,
- Olga Derbenevaa,
- Ryan Golhard,
- Pawel Golike,
- Sean O’Hearnf,
- Shawn Levyg,
- Prasanth Potluria,
- Maria Lvovaa,
- Antonio Davilaa,
- Chun Shi Lina,
- Juan Carlos Perinh,
- Eric F. Rappaporth,
- Hakon Hakonarsonc,
- Ian A. Trouncei,
- Vincent Procaccioj, and
- Douglas C. Wallacea,1
-
Contributed by Douglas C. Wallace, August 1, 2014 (sent for review May 15, 2014)
Significance
Mitochondria generate signals that regulate nuclear gene expression via retrograde signaling, but this phenomenon is rendered more complex by the quantitative differences in the percentage of mutant and normal mtDNAs that can exist within patient cells. This study demonstrates that depending upon its relative cytoplasmic levels, a single mtDNA point mutation can cause a discrete set of cellular transcriptional responses within cells of the same nuclear background. This qualitative regulation of nuclear gene expression by quantitative changes in mtDNA mutant levels challenges the traditional “single mutation–single disease” concept and provides an alternative perspective on the molecular basis of complex metabolic and degenerative diseases, cancer, and aging.
Abstract
Variation in the intracellular percentage of normal and mutant mitochondrial DNAs (mtDNA) (heteroplasmy) can be associated with phenotypic heterogeneity in mtDNA diseases. Individuals that inherit the common disease-causing mtDNA tRNALeu(UUR) 3243A>G mutation and harbor ∼10–30% 3243G mutant mtDNAs manifest diabetes and occasionally autism; individuals with ∼50–90% mutant mtDNAs manifest encephalomyopathies; and individuals with ∼90–100% mutant mtDNAs face perinatal lethality. To determine the basis of these abrupt phenotypic changes, we generated somatic cell cybrids harboring increasing levels of the 3243G mutant and analyzed the associated cellular phenotypes and nuclear DNA (nDNA) and mtDNA transcriptional profiles by RNA sequencing. Small increases in mutant mtDNAs caused relatively modest defects in oxidative capacity but resulted in sharp transitions in cellular phenotype and gene expression. Cybrids harboring 20–30% 3243G mtDNAs had reduced mtDNA mRNA levels, rounded mitochondria, and small cell size. Cybrids with 50–90% 3243G mtDNAs manifest induction of glycolytic genes, mitochondrial elongation, increased mtDNA mRNA levels, and alterations in expression of signal transduction, epigenomic regulatory, and neurodegenerative disease-associated genes. Finally, cybrids with 100% 3243G experienced reduced mtDNA transcripts, rounded mitochondria, and concomitant changes in nuclear gene expression. Thus, striking phase changes occurred in nDNA and mtDNA gene expression in response to the modest changes of the mtDNA 3243G mutant levels. Hence, a major factor in the phenotypic variation in heteroplasmic mtDNA mutations is the limited number of states that the nucleus can acquire in response to progressive changes in mitochondrial retrograde signaling.
Mutations in the 16.6 kilobase human mtDNA can cause a broad spectrum of multisystemic diseases (1, 2). Unlike chromosomal genes, which are present in only two copies per cell, the mtDNA can be present in hundreds to thousands of copies. If a cell acquires a deleterious mtDNA mutation, this creates an intracellular mixture of mutant and normal mtDNAs, a state known as “heteroplasmy.” Surprisingly, relatively subtle changes in the heteroplasmic levels can have dramatic effects on a patient’s phenotype (3), although the cellular basis for this phenomenon has remained unclear.
One of the most striking examples of this effect is observed with the mtDNA transfer RNA (tRNA)Leu(UUR) mutation at nucleotide 3243A>G, the most common human pathogenic mtDNA point mutation (4). The 3243A>G mutation perturbs mitochondrial protein synthesis resulting in amino acid misincorporation and electron transport chain deficiency (5). When this mutation is present at about 50–90% mtDNA heteroplasmy it can cause multisystem disease, the most recognizable phenotype known as the mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome (6). However, this same mutation at lower heteroplasmy levels has been associated with autism (7) and type I and type II diabetes in Eurasians (1, 8), and at very high levels can present as Leigh syndrome or perinatal lethality. MtDNA 3243A>G heteroplasmy also causes cell-type–specific biochemical effects depending upon the cell lineages (9). Given that different cell types differ primarily in their epigenomic context, one hypothesis as to why a continuous change in mitochondrial heteroplasmy could cause discrete phenotypic changes is that the cytoplasmic signal transduction systems and the nuclear epigenome may only be able to respond to changes in mitochondrial function in a limited number of discrete ways. This would create discontinuous changes in gene expression profiles and thus discrete clinical phenotypes (10).
To test this hypothesis, we generated transmitochondrial cytoplasmic hybrid cells (cybrids) with the same nuclear background that harbor increasing levels of the 3243A>G mtDNA mutation. We then compared the different cybrid lines for their cellular phenotypes, mitochondrial function, and transcriptome status. This comparison revealed striking nonlinear nuclear DNA and mtDNA transcriptome responses to progressively increasing mtDNA heteroplasmy levels, accompanied by phase-like changes in the transcriptional profile, presumably resulting from retrograde signaling effects on signal transduction and epigenomic pathways. This marked disconnect between mtDNA heteroplasmy and gene expression profile could explain the striking differences in clinical phenotypes of patients with different levels of the same mtDNA mutation.
Results
mtDNA Heteroplasmy Alters Mitochondrial Function, Structure, and Cell Size.
We transferred the wild-type (3243A) and mutant (3243G) mtDNAs from a heteroplasmic 3243A>G patient’s lymphoblastoid cell line into osteosarcoma 143B thymidine kinase-deficient (TK−) and mtDNA-deficient (ρo) cells and selected for transmitochondrial cybrids (Fig. 1A and SI Appendix, Fig. S1). Subsequent mtDNA depletion, reamplification, and cloning (11) resulted in a series of stable cybrids harboring ∼0%, 20%, 30%, 50%, 60%, 90%, and 100% 3243G mutant mtDNAs (SI Appendix, Fig. S1 and Extended Experimental Procedures Tables 1–3).
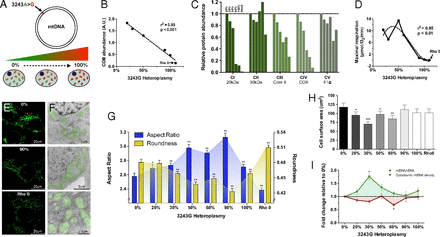
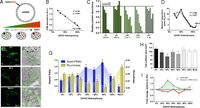
The mtDNA mutation 3243A>G alters cellular energetics, mitochondrial morphology, and cell size. (A) Experimental paradigm whereby increasing levels of heteroplasmy for the mtDNA mutation 3243A>G tRNALeu(UUR) were introduced into 143B(TK−) ρo cells. (B) Linear regression of mtDNA heteroplasmy and complex IV subunit COII protein levels. (C) Abundance of electron transport chain protein subunits of each complex (CI–CIV) probed by Western blotting of a constituent protein (20 kDa, 30 kDa, core II, COII, F1α) for complex I (CI), complex II (CII), complex III (CIII), complex IV (CIV), and complex V (CV) at different heteroplasmy levels. (D) Cybrid respiration rates fitting a nonlinear regression (third order polynomial) with maximal oxygen consumption revealing a threshold for respiratory deficiency above 60% 3243G heteroplasmy. (E) Confocal microscopy, and (F) electron microscopy (EM) representative images of 0%, 90% and ρo cell lines with pseudocolored (green) mitochondria. (G) Quantification of mitochondrial aspect ratio (length-to-width ratio) and roundness from confocal images. (H) Effect of heteroplasmy on cell size measured from electron micrographs and expressed as cross-sectional area, and (I) mtDNA copy number expressed as mtDNA/nDNA ratio (red line) and normalized to cell volume (green line). Significance is based on 95% (P < 0.05) or 99% (P < 0.01) confidence intervals (C.I.). Data for G and H are in means ± 95% C.I. and for I in means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA and Dunnett’s post hoc test.
The increasing levels of the 3243G mutation caused a dose-dependent reduction in mtDNA coded electron transport chain protein subunits (Fig. 1 B and C). However, maximum mitochondrial respiratory capacity was preserved beyond 60% heteroplasmy dropping to ρo levels at 90% 3243G mutant (Fig. 1D), this respiratory threshold being consistent with previous reports (12, 13). The retention of respiratory capacity in the presence of predominantly 3243G mutant mtDNAs is thought to result from the complementation of the mutant tRNALeu(UUR)s by wild-type mtDNAs tRNAs in trans due to mtDNA transcript mixing through mitochondrial fusion and fission (3, 14, 15).
Consistent with mitochondrial morphology being linked with mitochondrial bioenergetics (16, 17), and organelle fusion/elongation as an “early” mitochondrial stress response to electron transport chain defects (18), mitochondria in the 3243G cybrids became increasingly elongated as the percent mutant increased from 0% to 90%, but returned to near normal levels at 100% mutant mtDNAs (Figs. 1 E–G and 2). In contrast, the ρo cell’s mitochondria were maximally fragmented and swollen.

Representative electron micrographs of cybrid cell lines. Shown are lower magnification (Upper) and higher magnification (Lower) micrographs of cybrid cell lines harboring different percentages of 3243G heteroplasmy. M, mitochondria; N, nucleus; yellow arrowheads, endoplasmic reticulum.
Increasing mtDNA heteroplasmy also caused changes in mitochondrial ultrastructure, cristae and matrix electron density, and cytoplasmic ribosomal content (Fig. 2). In addition, compared with cells with only wild-type (3243A) mtDNAs, cells with 20% and 30% 3243G mutant mtDNAs showed a 19% and 40% reduction in cross-sectional area (Fig. 1H), corresponding to 25% and 54% reductions in cell volume, respectively (SI Appendix, Fig. S2D). Cell size then increased in the 50% 3243G cybrid to 80% of the normal volume, declined in the 60% 3243G cybrid to 60% of normal, and normalized in the 90–100% mutant and in ρo cells to 80–90% of normal volume (Fig. 1H). Changes in nuclear dimensions followed changes in cell size, resulting in a constant nuclear-to-cell ratio (SI Appendix, Fig. S2 B and C).
The cellular mtDNA copy number was determined for each 3243G heteroplasmy level. This revealed that the mtDNA/nuclear DNA (nDNA) ratio for the 20–30% 3243G cells tended to decline, returned to normal at 50%, declined significantly at 60% 3243G, and then returned to normal at 90–100% heteroplasmy (Fig. 1I). When mtDNA levels were normalized to cell volume, the mtDNA density within the various 3243G heteroplasmic cells was found to rise sharply from 0% through to 30% when it was 1.76-fold above the 0% cell line, followed by a progressive decline through 50%, 60%, and 90% 3243G heteroplasmy (Fig. 1I).
Effects on the Mitochondrial Transcriptome.
To determine the molecular basis of these physiological, morphological, and molecular changes, RNA was isolated from each cell line plus the ρo parent line, and following cytosolic rRNA depletion, converted to cDNA and high-throughput sequenced (RNA-Seq). Each cell line was found to have a distinct transcriptional profile involving clusters of related genes that increase or decrease coordinately (SI Appendix, Fig. S3).
Because the mutation occurred in the mtDNA tRNALeu(UUR) gene, we first examined the effects of increasing levels of the 3243G mutation on the expression of the mtDNA genes. The mtDNA encodes 37 genes: two rRNAs, 22 tRNAs, and 13 critical oxidative phosphorylation (OXPHOS) polypeptide mRNA genes (ND1–4, 4L, 5–6 for complex I; cytochrome b for complex III; COI–III for complex IV; and ATP6 and 8 for complex V) (Fig. 3A). The mtDNA is transcribed from three promoters, all located in the control region [white arc between the 12S rRNA and tRNA proline (P)] (Fig. 3A), one for the C-rich light (L) strand (inner circle), and two for the G-rich heavy (H) strand (outer circle). Both mtDNA strands are symmetrically transcribed into continuous polycistronic RNAs from which the mature rRNAs and mRNAs are freed by processing out the intervening tRNAs (1). In cells with only wild-type 3243A mtDNA, mtDNA transcripts accounted for 23.0% of the total cell’s transcriptome (Fig. 3B), as expected (19). However, when 20% of the mtDNAs harbored the 3243G mutation, the proportion of cellular transcripts of mtDNA origin dropped to 9.7%, and at 30% 3243G mtDNA transcripts constituted 12.7% of the total transcriptome. This is in similar proportion to the reduced cell volume. Because the decrement in mtDNA transcripts is twice that of the decrease in mtDNA levels in the 20–30% 3243G cybrids (Fig. 1I), this effect is not simply the result of reduced template abundance. Total cellular mtDNA transcript levels then increased to near normal levels in cells harboring 60–90% 3243G mutant mtDNAs, declining once more in cells with 100% 3243G mutant mtDNAs (Fig. 3B).
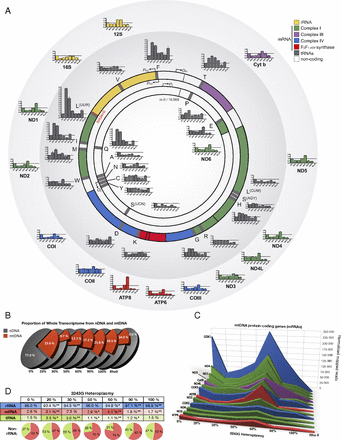
Regulation of the mitochondrial transcriptome in heteroplasmic cell lines. (A) Map of the mitochondrial genome and its 37 genes, showing origins of replication and promoters for the mtDNA light (L, inner) and heavy (H, outer) strands: replication origins OL and OH, and promotors PL, PH1, and PH2, respectively. Bar graphs show transcript levels of each gene at different heteroplasmy levels relative to 0% 3243G. Histograms are means ± SEM. (B) Relative abundance of nuclear- and mitochondrial-encoded transcripts in whole transcriptome analysis for each 3243G heteroplasmy level. (C) Expression level for the 13 mtDNA-encoded messenger RNA (mRNA) genes. (D) Relative abundance of mitochondrial ribosomal (rRNA), mRNA, and transfer RNA (tRNA) molecules with increasing heteroplasmy. *P < 0.01, **P < 0.001 relative to 0% by multiple two-tailed t tests. All data are from RNA sequencing, n = 3 runs per sample.
The expression of mtDNA mRNA genes was reduced in all heteroplasmic cells with the exception of the 60% 3243G cell line in which the mtDNA mRNA levels exceeded those of the wild-type cell line (Fig. 3C and SI Appendix, Figs. S4 and S5). The rise in the 60% 3243G cybrid mtDNA transcript levels is particularly striking because it changed in the opposite direction from the decline in mtDNA copy number and cell volume (Fig. 1I). However, the increased mtDNA transcripts was found to correlate with a sharp up-regulation of the mtDNA RNA polymerase (POLRMT) from 50% to 60% 3243G followed by a partial decline at 90% 3243G and a return to baseline at 100% 3243G mutant (SI Appendix, Fig. S4C). Hence, the increased mtDNA transcripts likely reflect increased mtDNA transcription and perhaps also mtDNA transcript stabilization.
The levels of the mtDNA tRNA genes exhibited considerable gene-to-gene variability (Fig. 3 A and D and SI Appendix, Figs. S4 and S5). The mutated tRNALeu(UUR) gene was selectively induced five- to eight-fold in the 20–30% heteroplasmy cells, with transcript levels of the other mtDNA tRNA genes being partially correlated with their location within the mtDNA (Fig. 3A). Hence, the ratios of mRNAs to tRNAs differed among the cybrids, with the tRNAs in the wild-type mtDNA-only cells (0% 3243G) consisting in 1.5% of the mitochondrial transcriptome, those in the 20–30% 3243G cell lines being 3.0–3.5%, those in the 50%, 60%, and 90% 3243G cell lines being 1.1–1.2%, and those in the 100% 3243G cell line returning to 1.5% (Fig. 3D). The up-regulation and down-regulation of tRNAs at low versus high mutation load, respectively, suggested a functional switch between 30% and 50% heteroplasmy.
Bioenergetic Adaptations to Increasing mtDNA Heteroplasmy.
To determine if the reduced mitochondrial gene expression triggered a compensatory induction of glycolysis, we analyzed the expression of all glycolytic genes in relation to the increasing levels of the 3243G mutant mtDNAs. In cells harboring 20–30% 3243G mtDNAs the expression levels of the glycolytic genes were similar to those of cells with only wild-type (0% 3243G) mtDNAs (Fig. 4A and SI Appendix, Fig. S6A). The striking reduction in cell size at 20–30% 3243G (Fig. 1H), the marked reduction in mtDNA gene expression in these cells, and the absence of compensatory up-regulation of glycolytic genes suggest that the 20–30% 3243G cells have a reduced cellular energy generating capacity.
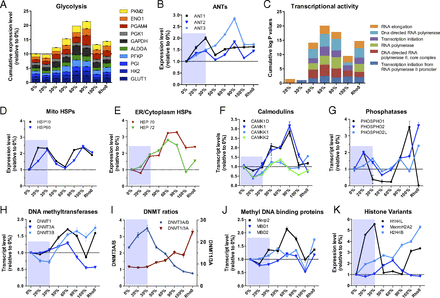
Biphasic induction of metabolic and stress response genes, signaling pathways, and of the epigenetic machinery. (A) Cumulative expression levels relative to 0% for genes encoding each enzymatic step of glycolysis in the various 3243G mutant cell lines. (B) Relative transcript levels for the adenine nucleotide translocator (ANT) isoforms. (C) Enrichment for gene ontology (GO) terms associated with gene families involved in transcriptional activity, expressed relative to 0% mutant. (D) Relative expression levels for the mitochondrial heat shock proteins (HSPs). (E) Expression levels of ER/cytoplasmic HSPs. (F) Expression levels of the Ca2+-sensitive signaling proteins calmodulin kinases (CAMK) and associated kinases (CAMKK). (G) Expression levels of cellular phosphatases showing inverse trend relative to kinases. (H) Expression of DNA methyltransferases (DNMTs). (I) Ratio of DNMT isoforms 3A/B and 1/3A. (J) Expression level of methyl-CpG binding protein 2 (Mecp2) and methyl-DNA binding domain (MBD1 and MBD2) genes. (K) Expression level of selected functional histone variants including the linker histone cluster 1 H4L (H1H4L); histone 2 variant MacroH2A2, which replaces conventional H2A in nucleosome and acts as transcriptional repressor; and histone cluster 2 variant H4B (H2H4B). Each gene is normalized to 1 in the 0% cell line and expressed in relative terms for cell lines of different heteroplasmy levels. Data for B and D–I are means ± SEM, n = 3 per cell line.
At 50% 3243G heteroplasmy, the expression of glycolytic genes rose reaching a maximum at 90% 3243G mutant mtDNA (Fig. 4A) with glycolytic enzyme genes being coordinately up-regulated (SI Appendix, Fig. S6A). The rise in glycolytic gene expression was associated with the decline in the synthesis and assembly of cytochrome c oxidase and its associated oxygen consumption (Fig. 1 B and C), a decline in cybrid growth rate, increased lactate production, and a decline in the relative maintenance of ATP levels (SI Appendix, Fig. S6 B–E).
The robust induction of glycolysis beginning around 50% heteroplasmy corresponded with a return toward normal cell size (Fig. 1H and SI Appendix, Fig. S2D), suggesting that the nucleus crossed an energetic threshold between 30% and 50% 3243G mutant, associated with a switch from a predominantly oxidative to glycolytic metabolism. When the 3243G mutant level reached 100%, glycolysis expression again declined and remained low in ρo cells, which do not have a functional respiratory chain (Fig. 4A). As a result, the 100% 3243G and ρo cell lines had significantly reduced ATP levels (SI Appendix, Fig. S6E).
The increased reliance on glycolysis of the 50–90% 3243G mtDNA cells corresponds with marked changes in the expression of mitochondrial inner membrane ADP/ATP transporters [adenine nucleotide translocator (ANT)] isoforms 1–3 (Fig. 4B). ANT1 mRNA increased from 0% to 20% and 30% 3243G mutant mtDNAs and then declined to a steady-state level in 50–100% 3243G mtDNA cybrids. ANT2 expression increased at 20–30% heteroplasmy, declined, and then peaked again at 90% 3243G heteroplasmy. ANT3 expression rose continuously from 0% to 90% 3243G heteroplasmy and then declined at 100% mutant. The ANT1 isoform is thought to be the most efficient at exporting mitochondrial ATP into the cytosol and hence is preferentially expressed in tissues with high ATP demand such as muscle, heart, and brain. This is consistent with ANT1’s up-regulation in the 20–30% 3243G cybrids, which may have been attempting to increase mitochondrial energy output, and its down-regulation in 50–100% 3243G cybrids, which had switched to glycolysis. The abrupt down-regulation in both ANT1 and ANT2 at 50% 3243G could explain the lower ATP levels in this cell line. ANT2 was the most highly expressed ANT isoform in all cell lines (SI Appendix, Fig. S6E), reaching its highest expression in the 90% 3243G cybrids, which also have the highest glycolysis levels. This is consistent with ANT2’s ability to import glycolytic ATP into the mitochondrion to support mitochondrial function during low OXPHOS ATP production (20). The striking induction of ANT3 was unprecedented.
Distinct Signal Transduction Systems Are Induced with a Functional Threshold Between 30% and 50% 3243A>G Heteroplasmy.
The changes in bioenergetic gene expression with increasing levels of the 3243G mutant mtDNA (Fig. 4 A and B) correspond to a striking and abrupt change in transcriptional regulation at the transition between 30% and 50% 3243G mutant. Compared with 0%, the 20–30% 3243G cell lines did not exhibit significant enrichment of genes classified by gene ontology (GO) as linked to transcriptional activity. However, in the 50–100% 3243G mutant cell there was an abrupt induction of genes involved in transcriptional regulation (Fig. 4C). Hence, there was a profound restructuring of the cellular transcriptome between 30% and 50% 3243G heteroplasmy.
Molecular chaperones of the heat shock protein (HSP) family were also differentially regulated. The mitochondrial HSP10 and HSP60 were up-regulated in 20–30% cybrids, declined to a minimum at 60%, and peaked again at 100% 3243G cybrids (Fig. 4D). In contrast, the endoplasmic reticulum (ER)/cytoplasmic HSP70 and HSP72 progressively increased in abundance starting at 20% 3243G, peaking at 90%, and declining again at 100% (Fig. 4E), reflecting compartment-specific unfolded protein stress responses. As the level of 3243G mtDNA increased, multiple antioxidant and redox regulatory systems also increased (SI Appendix, Fig. S7 A–C), in parallel with a rise in superoxide dismutase activity (SI Appendix, Fig. S7D). GO analysis also indicated a significant effect of mtDNA heteroplasmy on enrichment of differentially expressed families of genes involved in transcription, RNA splicing, autophagy/mitophagy, apoptosis, and neurodegenerative diseases (SI Appendix, Fig. S8).
The striking shifts in transcriptional activity were presumably initiated by mitochondrial metabolic retrograde signaling. These signals could include perturbation of mitochondrial Ca2+ homeostasis (21), altered mitochondrial reactive oxygen species (ROS) and reduction–oxidation (REDOX) signaling (10, 22), and altered mitochondrial production of reactive tricarboxylic acid (TCA) cycle intermediates such as α-ketoglutarate and other mitochondria-associated high energy compounds including ATP, acetyl-CoA, and S-adenosylmethionine (SAM) all of which are required to modulate signal transduction systems and the epigenome (10, 17).
Evidence that alterations in Ca2+ levels may be important in the observed 30% and 50% 3243G mutant transcriptional transition came from specific induction of the calmodulins, which were down-regulated at 20–30% 3243G followed by CAMK1D and CAMK1 induction from 50% to 90% 3243G mtDNAs, and then suppression at 100% 3243G mutant and in ρo cells (Fig. 3F). A reciprocal expression pattern was seen for several of the ubiquitous phosphatases, which perform opposite actions to CAMKs (Fig. 3G). In contrast to the coordinated transcriptional alterations, changes in the protein levels of various transcription factors and signal transduction proteins was less pronounced (SI Appendix, Fig. S9), presumably due to further translational and posttranslational regulation.
Evidence that changes in ROS production and REDOX balance contributed to the nuclear response to changes in mtDNA heteroplasmy is supported by the biphasic induction of the mitochondrial SOD2, which peaks at 20% and 90% 3243G, and the induction of SOD1 and peroxiredoxin 5 between 30% and 50% 3243G (SI Appendix, Fig. S7A). Evidence for REDOX regulation is seen in the biphasic induction of glutathione reductases 1 and 4 and the BolA-like 1 mRNA (SI Appendix, Fig. S7B) plus the progressive induction of the NADPH-generating enzymes isocitrate dehydrogenase 2 and malate dehydrogenase 2 between 50% and 90% 3243G mtDNAs (SI Appendix, Fig. S7C).
Evidence that metabolic signals may impact nuclear gene expression comes for alterations in the expression of genes that influence the nuclear epigenome in association with mitochondrial intermediates such as α-ketoglutarate, acetyl-CoA, and SAM. Altered regulation of DNA methylation was indicated by marked changes in the transcript levels of the known DNA methyltransferases DNMT1, DNMT3A, and DNMT3B (Fig. 4H). DNMT3B was down-regulated in cells with 20–30% 3243G mtDNAs, but at 50–90% 3243G both DNMT1 and DNMT3B rose dramatically, peaking at 60% and 90% mutant, respectively (Fig. 4H). The biphasic nature of the DNMTs’ response to the 3243G mutant level with the transition occurring between 30% and 50% can be illustrated by plotting the ratio of DNMT3A/3B, and DNMT1/3A (Fig. 4I). A related biphasic transcriptional pattern was also seen for the demethylation-related methylcytosine dioxygenase (TET3) (SI Appendix, Fig. S10A), an enzyme that uses α-ketoglutarate as a coreactant. Consistent with the mitochondrial 3243G mutation having an effect on methylation-related gene regulation, the expression of the disease-associated methyl DNA binding protein MeCP2 was induced at 30% 3243G mutant and peaked at 60% 3243G, falling again at 100% (Fig. 3J).
Because DNA methylation is a major cis-acting genetic regulatory mechanism (23) for gene networks (24), 3243A>G heteroplasmy must be having a direct effect on regulation of chromatin composition. This supposition is supported by changes in the expression of the histone variant genes. Linker histone cluster 1 H4L (H1H4L) was strongly induced in the 20–30% 3243G cells, dropped to baseline at 50–90% mutant, and then rose again in the 100% 3243G and ρo cells. In contrast, histone 2 variant macroH2A2, which replaces conventional H2A in the nucleosome and acts as transcriptional repressor, peaked at 60% 3243G mutant cells, whereas histone cluster 2 variant h4B (H2H4B) was induced continuously from 0% to 100% 3243G and ρo cells (Fig. 4K).
Further evidence for a phased change in nDNA gene expression was seen in the expression of histone deacetylases (HDACs) from 50% to 90% 3243G (SI Appendix, Fig. S10 B–E). Collectively, these changes were reflected in the cumulative enrichment of differentially expressed genes belonging to the chromatin remodeling pathways, which reached a maximum at 60% 3243G heteroplasmy (SI Appendix, Fig. S10F). As with the signal transduction proteins, changes in the transcription of epigenomic remodeler genes were more pronounced than were changes in the activity of epigenetic writers and of pangenomic abundance of specific posttranslational histone modifications (SI Appendix, Fig. S11).
Increasing mtDNA Heteroplasmy Induces Phase Transitions in Transcriptional Profiles.
To better define the transcriptional networks that were differentially activated in the progression from 0% to 100% 3243G heteroplasmy, we performed gene set enrichment analysis (GSEA) (25) and prepared a hierarchical clustering heat map showing enrichment of the functional pathways in each cybrid cell line. Again, relative to 0% 3243G control, striking discontinuities were observed between the 20–30%, 50–90%, and 100% 3243G mtDNA cybrids (Fig. 5A). In the 20–30% 3243G cybrids, the PI3 kinase-AKT (protein kinase B), the proto-oncogene myc, and mammalian target of rapamycin (mTOR) pathways were down-regulated and the epidermal growth factor (EGF), activating transcription factor 5 (ATF5), and bone morphogenic protein 2 (BMP2) gene targets were up-regulated. In the 50–90% 3243G cybrids the forkhead box P3 (FOXP3), transcription factor 21 (TCF21), and signal transducer and activator of transcription 3 (STAT3) pathways were down-regulated, whereas the hypoxia inducible factor 1 (HIF1), myc, Kirsten rat sarcoma viral oncogene homolog (K-Ras), peroxisone proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), and “aging” and “senescence” pathways were up-regulated. At 100% 3243G the interferon beta (Ifn-β), polycomb repressive complex 2 (PRC2), STAT3, specifying protein 1 (SP1), and ETS-related gene 1 pathways (EGR1) were down-regulated (Fig. 5A).
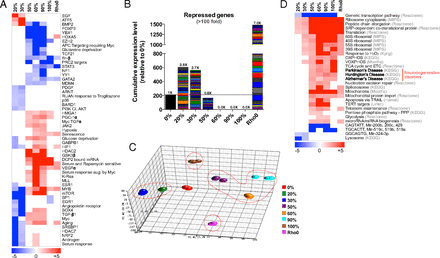
Multiphasic reprogramming of nuclear gene expression. (A) Gene set enrichment analysis (GSEA) of transcriptional and signaling pathways from whole transcriptome data showing differential gene transcription phases of cybrids in the heteroplasmy ranges of 0%, 20–30%, 50–90%, and 100% 3243G plus ρo cells (log P values relative to 0% cell line). (B) Expression levels of 211 genes that are up-regulated at 20–30% 3243G but strongly repressed >100-fold at 60–100% 3243G, enriched for transmembrane and G-protein–coupled receptor systems (see SI Appendix, Fig. S12 for details). (C) Principal component analysis (PCA) showing the striking discontinuity in gene expression profiles of 0% (normal), 20–30% (diabetes and autism), 50–90% (degenerative diseases), 100% (perinatal diseases and Leigh syndrome), and ρo (mitochondrial null). (D) Pathway analysis comparing the 3243G cybrid transcript profiles to those from public databases (databases shown in gray), log P significance values.
In analyzing differential gene expression profiles, we observed 211 genes that were induced in the 20–30% cybrids, but were repressed more than 100-fold in 60–100% 3243G cybrids (Fig. 5B). Functional classification analysis (Database for Annotation, Visualization, and Integrated Discovery v6.7, DAVID) revealed that these genes predominantly encompassed transmembrane proteins and signal transduction elements including the G-protein–coupled receptors systems (SI Appendix, Fig. S12).
The distinction between the gene expression profiles of 0%, 20–30%, 50–90%, and 100% 3243G heteroplasmy cells plus the ρo cells was further demonstrated when the whole transcriptome data were analyzed by principle component analysis (PCA). The 20–30% cybrids and the 50–90% cybrids occupy diametrically opposed transcriptional spaces (circled), with the 0% mutant cybrids falling between these clusters (Fig. 5C). The 100% 3243G and the ρo cells also occupy totally distinct spaces, underscoring their widespread transcriptional differences with the other cybrid lines.
To understand which functional pathways distinguish these clusters, we interrogated publicly available databases for similar differential genome-wide expression profiles. This analysis further highlighted the difference between 20–30% and 50–90% 3243G, and to a lesser extent between 50–90% and 100% 3243G cybrids. Notably, the cybrids with 50–90% 3243G exhibited significant up-regulation of mitochondrial function (OXPHOS, TCA, etc.) genes (Fig. 5D), consistent with the up-regulation of PGC-1α at high mutation levels (26) and in association with increased ribosomal biogenesis and telomere regulation (Fig. 5A). The induction of these genes contrasted sharply with the marked down-regulation of CAGTATT and GGCAGTG micro-RNA gene expression. Lastly, the gene expression profile of the 50–90% 3243G cybrids was strikingly reminiscent of those reported for neurodegenerative diseases such as Alzheimer, Parkinson, and Huntington diseases (Fig. 5D). Therefore, the gene expression profiles associated with mtDNA 3243G mutant retrograde signaling fall into four distinct transcriptional phases associated with 0, 20–30, 50–90, and 100% 3243G mutant levels.
Discussion
Our results demonstrate that continuous changes in mtDNA heteroplasmy result in discontinuous remodeling of nuclear DNA and mtDNA gene expression profiles due to alterations in both the signal transduction and epigenetic regulatory processes. Whereas the traditional medical view is that human genetic mutations result in predictive phenotypes according to the rules of Mendelian genetics, our study reveals that variation in the percentage of a single mtDNA heteroplasmic nucleotide substitution can lead to a multiplicity of cellular phenotypes. This occurs through phase-like alterations in gene expression profiles and carries profound implications for the biochemical and molecular basis of age-related diseases and aging (27).
That mitochondrial dysfunction can influence nuclear gene expression has been demonstrated in yeast (28), nematode (29), and mammalian systems (30). Existing studies indicate that mitochondrial signaling can trigger both adaptive and maladaptive cellular responses, with a possible pivotal factor being the degree of mitochondrial dysfunction. However, no previous study has examined the dose–response dynamics of transcriptional and cellular adaptive responses to the full range of mtDNA heteroplasmy in human cells.
Heteroplasmy levels are known to vary between the different tissues of individuals (31), which is one explanation offered for the clinical variability seen in mtDNA disease (9). However, the present study demonstrates that variation in mtDNA heteroplasmy levels can also differentially activate various cellular metabolic pathways. Because the regulation of energy metabolism must predate tissue differentiation many of the regulatory responses to changes in 3243G levels seen in 143B(TK−) cells are likely to be pertinent to a range of tissues.
The striking difference in mtDNA transcript levels between 20–30% and 50–90% 3243G cell lines corresponds to the transition seen between patients presenting with diabetes and autism at 20–30% 3243G heteroplasmy and neuromuscular disease at 50–90% 3243G mutant (SI Appendix, Fig. S13). The reduced mtDNA transcripts in the 20–30% 3243G diabetes range (8) along with the reduced cell size and increased mtDNA density are consistent with the conclusion that low levels of mtDNA heteroplasmy cause significant decreases in mitochondrial function without compensatory up-regulation of glycolysis. The apparent resulting deficiency in cellular energy production provides a potential explanation for the consistent observation that type II diabetes is associated with a partial defect in mitochondrial bioenergetics (32). At 50–90% 3243G levels, glycolysis is induced to partially compensate for the increased OXPHOS defect. This is also associated with the induction of the suite of genes that have been associated with neurodegenerative diseases such as Alzheimer and Parkinson diseases (see Fig. 5D).
Although it might be argued that the bioenergetic changes seen in this osteosarcoma cell model may have little relevance to differentiated tissues due to the heavy utilization by cancer cells of glycolysis, the Warburg effect, more recent data demonstrate that cancer cells are not OXPHOS deficient but rather have increased glycolysis to meet biosynthetic requirements (33). Hence the relative changes in mitochondrial and glycolytic metabolism associated with changes in mtDNA heteroplasmy observed here could well reflect changes in gene expression that occur in noncancer cells.
In response to the changes in OXPHOS energy production between 20–30% and 50–90% 3243G mutant, the global gene expression profiles of cybrids changes abruptly. At low levels heteroplasmy (20–30% 3243G) the transcriptional signatures include the down-regulation of mTOR signaling and other growth pathways (Fig. 5A and SI Appendix, Fig. S9), the overall reduction of mtDNA gene expression and induction of mitochondrial chaperones, and an associated reduction in cell size. In contrast, at high-level heteroplasmy (50–90% 3243G) the transcriptional profile includes the up-regulation of pathways related to both glycolysis and mitochondrial functions including OXPHOS, the down-regulation of genes involved in transmembrane signal transduction, and the engagement of expression profiles associated with senescence and telomere maintenance. These differences could represent alternative cellular strategies for adapting to changes in energy availability, with different adaptive strategies used, depending on the length and extent of the reduced mitochondrial energy output. During transient periods of low energy resource availability recapitulated by the effects of 20–30% 3243G heteroplasmy, the programmed nuclear response may be to conserve energy through reduced energy utilization and increased recovery of cellular energy resources. However, with prolonged periods of more severe energy deficiency, recapitulated by the effects of 50–90% 3243G, the programmed nuclear response may be to coordinately up-regulate all energy-generating systems to maximize the exploitation of the available caloric resources. In contrast to the low energy deprivation state, the high energy deprivation state would require a major restructuring of many cellular functions and associated gene expression profiles, which appears to be common to many degenerative diseases and aging (27). Irrespective of the evolutionary arguments, it is clear from the PCA of global gene expression changes that graded changes in mitochondrial energy output result in quantized changes in gene expression profile (Fig. 5C) and that the 50–90% 3243G expression profile mimics those associated with Alzheimer, Parkinson, and Huntington diseases, thus supporting the conclusion that these neurodegenerative diseases have an underlying bioenergetic etiology.
Whereas alterations in mitochondrial Ca2+ (33), ROS, and REDOX (34) regulation are established mechanisms for mediating mitochondrial retrograde signaling, the current study also indicates the importance of mitochondrial alterations in chromatin remodeling presumably through mitochondrially generated metabolic intermediates (10, 33). This can be inferred by changes in the expression of genes encoding the DNA methylation machinery. DNMT1 and 3A were stable or down-regulated at 20–30% 3243G, but were coordinately up-regulated at 50–90% 3243G. Likewise, the methyl DNA binding protein, MeCP2, was strongly induced at higher heteroplasmy levels, peaking at 60% 3243G mutant. The DNMT isoforms, which have nonoverlapping gene-specific DNA methylation activities (35), have been reported to be differentially associated with mitochondrial function. DNMT3A is concentrated in synaptic boutons within synaptic mitochondria (36) and missense mutations in the DNMT3A gene have been observed in a patient with autism (37). DNMT1 has been located in fibroblast mitochondria (38) and mutations in the DNMT1 gene have been linked to sensory neuropathy, dementia, and deafness (39). Mutations in the MeCP2 gene cause Rett syndrome, a syndromic autism spectrum disorder (40), and Rett syndrome has been associated with abnormal brain mitochondrial structure and function (41, 42). Thus, the regulation of these epigenetic regulators by mtDNA heteroplasmy could cause broad transcriptional effects of clinical significance.
One technical concern with this study is that 143B(TK−) cells were derived from an osteosarcoma, and this and other cancer cell lines have been found to be karyotypically unstable and prone to alterations in gene expression (43) and bioenergetics function (44) during cybridization, removal of mtDNA, and propagation in different culture conditions. There are several reasons why we feel that it is unlikely that variation in the 143B(TK−) cybrid nuclei could account for the variation that we observed in gene expression profiles of the mtDNA 3243G mutant cybrids.
The first possibility is that the transcriptional phase changes seen in the 0%, 20–30%, 50–90%, and 100% 3243G cybrids are the result of random alterations in the 143B(TK−) nDNA, resulting in random changes in the transcription profile. This possibility is unlikely because changes in the expression of many cybrid nDNA genes are nonrandom in relation to the 3243G mutant mtDNA levels. For example, the expression of catalase (SI Appendix, Fig. S7A), HSP70, and HSP72 (Fig. 4E), and alternate histone H2H4B (Fig. 4K) all increased incrementally from 0% to 90–100% 3243G in progressive continuous curves. Furthermore, the expression of genes that displayed the phase differences in gene expression showed similar levels of expression as seen in other cell lines with similar heteroplasmy levels for both the 20–30% and 50–90% cybrid clusters. Random changes in gene expression would not show either of these correlations with the 3243G mutant levels.
The second possibility is that the cybrids in each transcriptional phase could be clonally derived from the same founder cell and that the different clones harbored nDNA variants that created the phase changes. For this to be true, the cybrids in each of the phased heteroplasmy classes (0%, 20–30%, 50–90%, 100%, or ρo cells lines) would have to have been derived from separate founder 143B(TK−) clones. This possibility can be excluded for two reasons. In one, we used gene fusions as markers to show that the nuclei of the cybrids within the same transcriptional phase were not derived from the same 143B(TK−) nuclear clones. Gene fusions were detected through transcripts that incorporated two noncontiguous gene sequences, presumably representing the products of intra- or interchromosomal rearrangements resulting in gene fusions. Multiple gene fusion transcripts appear in the various cybrids and several of these are shared between the different cybrid clones, making them useful markers for the nuclear lineages of the cybrids (SI Appendix, Fig. S14). However, no correlation was found between the array of gene fusion events in the different cybrid nuclei and the transcriptional phases of the cybrids (0%, 20–30%, 50–90%, 100% heteroplasmy and ρo cells) (SI Appendix, Fig. S14). The second set of data that excludes the 143B(TK−) nuclear clonal explanation for the phase changes came from our sequencing of the mtDNAs of the different cybrid clones (SI Appendix, Extended Experimental Procedures). This revealed a de novo synonymous mtDNA mutation at nt 9494A>G in the COIII gene in two cybrid lines, 30% and 50% 3243G. Because these two clones shared a common de novo mutation, they must have been derived from a common ancestral 143B(TK−) clone; yet these two cybrids fall into the two most distinct transcriptional gene expression phases. Therefore, cybrids with similar expression phases can have different clonal origins and cybrids with different expression phases can have the same clonal origin, thus excluding the possibility that the expression phases are the result of clonal origins of the 143B(TK−) nuclei.
The third possibility is that unique features of the 143B(TK−) osteosarcoma nucleus cause the observed transcriptional phase changes. To test this possibility, we transferred the 3243G heteroplasmic mtDNAs from selected 143B(TK−) cybrids into the SH-SY5Y neuroblastoma nuclear environment (13). This was accomplished by inactivating the mitochondrial donor 143B(TK−) cybrid nuclei with actinomycin D (45) and the SH-SY5Y mitochondria with rhodamine 6G (46), cell fusion, and clonal isolation. SH-SY5Y cybrid clones were produced harboring 0%, 70%, and 100% 3243G mutant mtDNAs and the physiology of these cell lines (13) compared with the gene expression profiles of the 143B(TK−) cybrids. The SH-SY5Y cybrids showed a 3243G dose-dependent increase in glucose consumption and lactate production, consistent with the induction of glycolysis at high 3243G levels in the 143B(TK−) cybrids. Furthermore, the SH-SY5Y cybrids showed a strong up-regulation of mitochondrial respiration at 70% 3243G mutant (13), which corresponds with the maximum induction of mtDNA mRNAs and of POLRMT in the 60% 3243G 143B(TK−) cybrids (Fig. 3C and SI Appendix, Fig. S4 C and D). Finally, cybrids on both nuclear backgrounds with high 3243G levels exhibited evidence of increased oxidative stress through either elevated ROS production or increased antioxidant enzymes.
Hence, we can exclude the possibility that the observed differential phased gene expression of the 20–30% and 50–90% 3243G cybrids is the result of clonal differences in the 143B(TK−) nuclei. Rather, the phase changes must be the result of differences in the levels of the mtDNA 3243G mutant.
In conclusion, our data demonstrate that subtle changes in mtDNA heteroplasmic genotype can have profound effects on the nuclear gene expression state. This retrograde signaling likely involves mitochondrially mediated changes in Ca2+, ROS and REDOX, and levels of reactive mitochondrial metabolic intermediates. This implies that the stochastic and quantitative genetics of the mtDNA may be a major contributor to the seeming genetic “complexity” of the common metabolic and degenerative diseases and aging.
Materials and Methods
Generation of Transmitochondrial Cybrids.
A mtDNA-deficient (ρo) cell line derivative from the osteosarcoma 143B thymidine kinase-deficient (TK−) cell line [143B(TK−), American Type Culture Collection CRL 8303] (47) was fused to enucleated lymphoblastoid cells (47) from a patient heteroplasmic for the tRNALeu(UUR) nucleotide (nt) 3243A>G mtDNA mutation (48) using polyethylene glycol (47). After being established, cybrids were selected and isolated. Heteroplasmy for the tRNALeu(UUR) nt 3243A>G mtDNA mutation was monitored by restriction fragment length polymorphisms (RFLP), and the whole mtDNA genome sequenced by next-generation sequencing at different passages. A series of remarkably stable clones was ultimately obtained harboring ∼0%, 20%, 30%, 50%, 60%, 90%, and 100% 3243G mutant mtDNA. See SI Appendix for complete details.
Mitochondrial Composition and Function.
Electron transport chain (ETC) subunits complex I 20 kDa, complex II 30 kDa, complex III core II, complex IV COII, and complex V (ATP synthase) F1α abundance were quantified by Western blotting, and mitochondrial respiratory complexes were separated by Blue Native-PAGE (49). Mitochondrial respiration in each 3243G heteroplasmic cell line was measured using the Oxygen Biosensor System (BD Biosciences). Cellular ATP was measured by bioluminescence normalized to protein content. MtDNA copy number was determined by quantitative real-time PCR (qRT-PCR) by the ΔΔCt method.
Electron and Confocal Microscopy.
Cells in suspension were fixed with 2.5% (vol/vol) glutaraldehyde, 2.0% (vol/vol) paraformaldehyde in 0.1 M sodium cacodylate buffer, postfixed and embedded to produce ultrathin sections imaged by transmission electron microscopy. For quantification of cell and nuclear size, cell and nuclear contours were manually traced in ImageJ by two different investigators blinded to group identity. To normalize mtDNA content to cell content, cell volume was then calculated assuming spherical shape (cells in suspension). For confocal microscopy, cells were stained with Mitotracker Green TM and imaged using a laser scanning confocal microscope in a air- and temperature-controlled, humidified chamber. Mitochondrial morphology was quantified using an automatic segmentation macro and standard shape factors.
RNA Sequencing and Library Preparation.
Total RNA was extracted, depleted of cytosolic rRNA, quality checked, and quantified before library preparation. Barcoded samples in triplicates from eight experimental groups were sequenced concurrently on the ABI SOLiD 5500 platform using paired-end chemistry of 50 (forward) × 35 (reverse) base pairs. Sequenced reads were mapped using the whole-transcriptome paired-end mapping pipeline against the 1000 Genomes Build 37 Decoy 5 reference (50) with default parameters. Mapped reads were further filtered postmapping to retain only those reads in which both read pairs from each paired-end fragment were properly paired, resulting on average in 114 million reads per replicate.
RNA sequencing data were further analyzed using Partek Genomics Suite, normalized, and expressed as reads per kilobase per million reads (RPKM), with R package DESeq yielding similar results. DAVID (51) was used to derive functional significance from highly repressed genes. To quantify alterations in gene expression between samples with different mtDNA heteroplasmy, the sample with 0% heteroplasmy was set as baseline, and fold change with all other heteroplasmy level genes ranked from most up-regulated to most down-regulated. GSEA was conducted (25) with the gene sets from the Molecular Signatures Database (MolSigDB v4.0), with a false discovery rate (FDR) threshold of <0.1. Hypergeometric distributions were used to evaluate the significance of enriched biological processes using the GO categories from differentially regulated gene lists. Only biological processes with significant enrichment (P < 0.01) were selected and visualized by heatmap.
Chromatin Remodeling and Enzymatic Assays.
All assays were performed on either nondenatured nuclear fractions or total cellular extracts using Western blotting or ELISAs according to the manufacturers’ instructions.
Please refer to SI Appendix, Extended Experimental Procedures for complete details, including antibodies, primer sequences, RNA-sequencing, bioinformatics, and cell culture conditions.
Acknowledgments
The authors thank Katelyn Sweeney and the University of Pennsylvania EM Core Facilities for technical assistance on parts of this project. This work was supported by Simon Foundation Grant 205844 and by National Institutes of Health Grants NS21328, NS070298, AG24373, and DK73691 (to D.C.W.). M.P. is supported by a postdoctoral fellowship from the Canadian Institute of Health Research Institute of Neuroscience as part of the Canadian Epigenetics, Environment, and Health Research Consortium.
Footnotes
- ↵1To whom correspondence should be addressed. Email: wallaced1@email.chop.edu.
-
Author contributions: D.C.W. designed research; M.P., S.H., O.D., R.G., P.G., S.O., S.L., P.P., M.L., A.D., C.S.L., E.F.R., I.A.T., V.P., and D.C.W. performed research; M.P., J.Z., S.H., R.G., J.C.P., E.F.R., H.H., V.P., and D.C.W. analyzed data; and M.P., V.P., and D.C.W. wrote the paper.
-
The authors declare no conflict of interest.
-
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE56158).
-
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1414028111/-/DCSupplemental.
Freely available online through the PNAS open access option.